Bone sarcomas are rare and account for only 0.2% of all neoplasms. They represent a group of cancers that occur predominantly in children and young adults. Intrinsic to their aggressive behaviour these tumours are lethal in about 40% of patients despite modern multimodality therapy. Although substantial progress is made over the past 10 years in understanding these tumours at the biological, pathologic and genetic level, this has not been translated to more effective therapies so far. Within the group of bone tumours a large number of histologically, clinically and genetically distinct tumour entities are discerned.
The 2002 WHO classification1, in which for the first time genetic and histological features are integrated, recognises 32 different entities.
Since bone tumours as a group are already rare, this creates even a bigger problem in reaching significant numbers studying the different types of bone tumours. Research is performed on these rare and complex tumours in relatively small research groups, which are inherently hampered by the lack of availability of substantial numbers of cases as well as a critical technical and/or multidisciplinary mass.
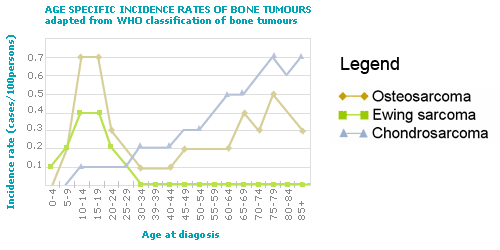
As can be seen below the age distribution of two of the most common types of bone tumours, Ewing sarcoma and osteosarcoma shows that this is primarily a problem in children and young adults.
The second peak of osteosarcoma incidence at later age is considered as a result of a predisposing condition, e.g. radiation. Chondrosarcoma incidence increases with age, however precursor lesions are mainly found in children and adolescents.
Therefore, depending on the researched tumour group the target population will vary. In case of osteosarcomas and Ewing sarcomas the main target populations will be that of children, whereas in case of the chondrosarcomas the main target population will be the adults. |